New development of ab initio calculation methods
Ab initio multi component molecular orbital (MC_MO) method
We have extended the concept of molecular orbital to the motion of proton or positron.
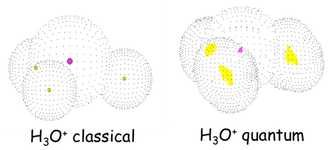
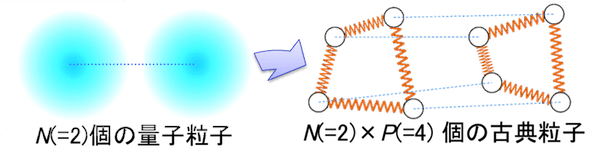
Ab initio path integral simulation (PI) method
We have achieved full quantum treatment of molecular systems by using ab initio method and path integral technique for electron and nuclei, respectively.
Ab initio multi component quantum Monte Carlo (MC_QMC) method
We have also extended quantum Monte Carlo (QMC) method, one of the most accurate ab initio method, to the multicomponent systems.
